I’m working with the Velvet Assembler as part of my virus identification project. When I’m not trying to write a perl module to complement the already-bulky script files that I’m working with, I like to do something that I usually wish most other bioinformatics scientists I’ve met would do as well: I like to delve into the mathematics behind the program.
Velvet Assembler constructs de Brujin graphs in a manner like so:
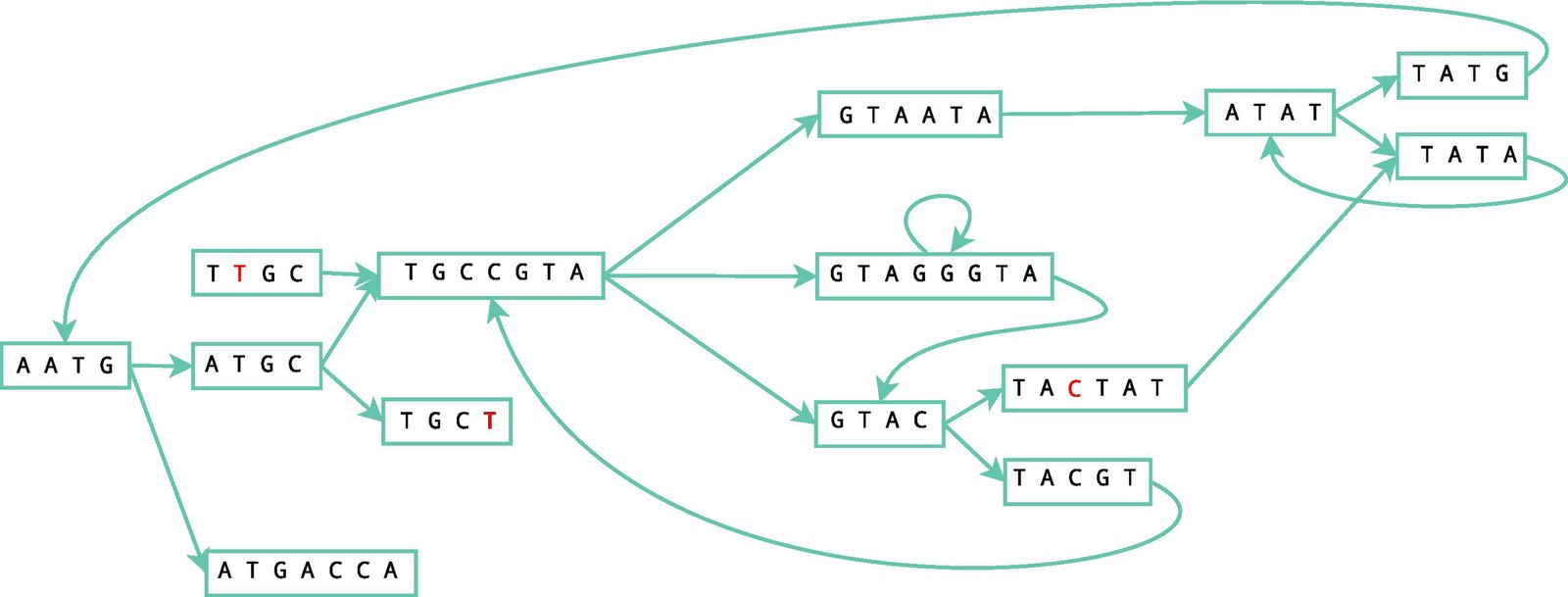 |
| source |
Confusing, right? Velvet uses the separated small reads to make overlapping regions that be analyzed for errors and compressed for simplicity. These small reads are constructed into a de Brujin graph, which contains a list of all the overlapping segments. See those arrows in the picture above? Those are edges. And the rectangles are our nodes. We have to tell the computer the rules of how to play the game of read alignment before it can go crazy in the sandbox, so we’ve decided on a directed graph that can follow certain parameters. When the ending of one region connects with another, it forms a “bubble” that can be corrected using the Tour Bus algorithm, which is similar to the Dijkstra’s algorithm, which we’ll get to in another post.
And that’s basically all I need to know about it for my research project. But, as with most mathematical oddities, that’s not as much as I want to know.
With the help of my friend, Rosalind, I was introduced to de Brujin graphs. To construct one, we could easily write a program that iterates through a list of k-mers from each given segment and, then, record the ones that overlap by all but one of their base pairs. (My solution for the Rosalind problem can be found here, by the way.) This raises a lot of questions about how to reconstruct an entire genome from the smaller segments. The favorite problem of every computer scientist, the Traveling Salesman Problem, (and in some cases, something a bit more unique) arises when we have to decide what is the best way to connect each smaller segment into a larger picture.
I hope to optimize the Velvet Assembler portion of the Virus Detection program with a greater understanding of the mathematics behind it. So far, it seems that all my work simply relies on my ability to use different commands and put them in script files. And, recently, I’ve been bogged down with error message after error message in the implementation of a self-written subroutine module. Sometimes you need to relax with some puzzles. Even if the pieces are all so big that they overlap with one another.

Leave a comment